EEG Spikes in Young Children: Benign Variants, Hidden Seizures, and Treatment Insights
Introduction
When an electroencephalogram (EEG) on a young child shows frequent epileptiform spikes – even in the absence of obvious convulsions – it raises challenging questions. Are these EEG abnormalities part of a benign, self-limited pattern, or do they signal an epileptic encephalopathy that could silently harm the child’s development? What other conditions could cause such EEG findings, and how do clinicians distinguish them from syndromes like Landau–Kleffner syndrome (LKS) or electrical status epilepticus in sleep (ESES)? Most importantly, if a child has no visible seizures but clear spike-wave discharges on EEG, should treatment (such as anti-seizure medications) be considered? This article delves into these questions, examining the nature and causes of EEG spikes in children, the spectrum from benign to severe epileptic syndromes, signs of “hidden” seizures that parents should watch for, and evidence on whether treating subclinical epileptic activity can improve developmental outcomes. We also explore how advanced technologies – including artificial intelligence (AI) and large language models (LLMs) – may aid in diagnosing these conditions and guiding therapy.

Understanding EEG Spikes and Benign vs. Pathological Patterns
EEG spikes (also called interictal epileptiform discharges) are transient waveforms that stand out from the normal brain background activity. They indicate a momentary, hypersynchronous firing of neurons, reflecting cortical hyperexcitability and the presence of an underlying epileptic network. In practical terms, seeing spikes or spike-and-wave complexes on EEG means that the brain has an increased potential for seizures. However, it’s crucial to note that epileptiform activity on EEG alone is not sufficient to diagnose epilepsy in a child with no clinical seizures. Some children – even otherwise healthy ones – can have epileptiform discharges incidentally, without ever having observable seizures. In such cases, the EEG abnormality represents a risk or tendency, but not a definite disease unless seizures or developmental regressions occur.
There are well-recognized benign EEG variants in children. For example, benign “rolandic” spikes (centrotemporal spikes) are seen in self-limited epilepsy with centrotemporal spikes (SeLECTS), previously known as benign rolandic epilepsy. This syndrome typically affects children around age 3–12 and is considered idiopathic (no structural brain lesion). The hallmark is high-amplitude spikes in the centrotemporal regions, especially activated during drowsiness and sleep. Children with this EEG pattern often have infrequent, brief focal seizures (often involving facial twitching or speech arrest at night) or sometimes no noticeable seizures at all. Importantly, their neurological and cognitive development is usually normal. In fact, the long-term prognosis in classical rolandic epilepsy is invariably excellent, with nearly all children outgrowing the seizures by adolescence and no lasting neurological deficits. Follow-up studies show that most incidental epileptiform discharges in otherwise healthy children will disappear spontaneously as the brain matures. This suggests that benign childhood spikes are related to a transient, mild disruption in brain maturation that resolves over time. Consistent with this, genetic studies of rolandic epilepsy have found no major mutations but point to hereditary factors – for example, a susceptibility locus on chromosome 15q14 – and a hypothesized “mild and reversible impairment of brain cortical maturation” underlying the spikes.
Not all EEG spike patterns are benign, of course. At the severe end of the spectrum are the epileptic encephalopathies, where ongoing epileptiform activity actively interferes with brain development. LKS and ESES/CSWS (continuous spike-and-wave during slow-wave sleep) are prime examples. In Landau–Kleffner syndrome (LKS) – a rare childhood epilepsy syndrome – previously normal children lose language skills (aphasia), either suddenly or progressively, and their EEG shows frequent epileptic spikes, often maximally over the temporal lobes, especially during sleep. About 70–85% of LKS patients have clinical seizures (which can be mild), but the defining feature is the “acquired epileptic aphasia,” i.e. the child can no longer understand or express speech due to the epileptic activity disrupting language networks. By definition, LKS requires the clinical pattern (language regression) and a characteristic EEG showing paroxysmal epileptiform activity, often a form of electrical status in sleep. The causes of LKS are not fully understood – some cases appear related to an autoimmune process, and others have been linked to mutations in the gene GRIN2A, which is involved in neural signaling. (Interestingly, GRIN2A mutations are found in a subset of children with the broader epilepsy-aphasia spectrum, which includes LKS and related syndromes, but not everyone with such mutations develops LKS.)
Another related condition is Epileptic Encephalopathy with Continuous Spike-and-Wave during Sleep (CSWS), also known as ESES when referring to the EEG pattern. Here the EEG during non-REM sleep is occupied by nearly continuous 1–3 Hz spike-wave discharges – sometimes 85% or more of the slow-wave sleep time. Clinically, CSWS/ESES leads to global or selective neurocognitive regression: children may lose academic skills, exhibit behavioral deterioration, or develop motor deficits like ataxia or speech problems, typically between ages ~4 and 8. Seizures are usually present (often infrequent and occurring in sleep, such as atypical absences or focal motor seizures), but the distinctive feature is that the persistent spikes during sleep are thought to be a major driver of the cognitive impairment – a classic epileptic encephalopathy where “the epileptiform EEG abnormalities themselves contribute to a progressive disturbance in cerebral function”. ESES/CSWS is often an age-limited phenomenon: the intense spike activity usually remits spontaneously by puberty (around 8–12 years of age). Unfortunately, even after the EEG returns to normal, many children are left with permanent learning or language disabilities from the years of nocturnal epileptic activity. This makes early recognition and treatment of ESES critical. ESES can occur idiopathically (in children with previously normal development and no lesion) or in those with underlying brain abnormalities. In roughly half of cases, a cause like a subtle structural lesion is identified – for example, perinatal stroke scars or focal cortical dysplasia – often involving the thalamus or perisylvian regions important for sleep networks. In the other ~50%, MRI is negative, implying that the cause is a network-level dysfunction without a visible lesion. In those idiopathic cases, researchers suspect a functional disturbance of the thalamo-cortical circuits that generate sleep spindles; essentially, the normal sleep rhythms are hijacked and replaced by pathological spike-waves, wreaking havoc on memory and learning consolidation.
Between the benign and encephalopathic extremes lies a spectrum of intermediate possibilities. Some children with so-called “benign” focal spikes can have atypical evolutions, where seizures become more frequent and developmental problems emerge. For instance, a child initially diagnosed with benign rolandic epilepsy might later develop learning difficulties or more diffuse spike activity – an evolution toward the CSWS end of the spectrum (sometimes termed atypical benign partial epilepsy or atypical rolandic epilepsy). Conversely, not every child with language delay and spikes has true LKS; many may have an autism spectrum disorder or developmental language disorder with coincidental EEG abnormalities. Distinguishing these scenarios requires careful clinical correlation: LKS typically involves a previously normally developing child who loses language (auditory verbal agnosia and expressive aphasia), whereas a child with autism or congenital language delay might never have acquired speech in the first place. The EEG in LKS often shows a profusion of spikes in sleep (sometimes meeting ESES criteria), predominantly in language-related cortical areas, whereas in non-LKS autism the spikes might be more scattered or less intense. Both groups can show normal MRI(since LKS is not usually due to a visible lesion either). Thus, the context and evolution (regression vs. delayed development) help differentiate them, even if the EEG patterns overlap.
Hidden Seizures and Subtle Signs Parents Should Watch For
One of the biggest concerns with EEG spikes in young children is the possibility of “hidden” seizures – seizures so subtle that they go unrecognized as seizures. Especially in toddlers and non-verbal children, epileptic episodes may not manifest as dramatic convulsions. Instead, a child might exhibit brief, odd behaviors or lapses that a parent could easily attribute to daydreaming or quirkiness. Mothers and fathers should be vigilant for certain patterns in day-to-day behavior that could indicate seizure activity:
- Unresponsive staring spells: A child may suddenly pause and stare blankly into space, not reacting to their name or touch, for a few seconds at a time. If the child is truly non-responsive during these episodes (and doesn’t recall them), they could be absence seizures rather than ordinary daydreaming.
- Repetitive automatic behaviors: The child might perform a repetitive motion – for example, lip-smacking, picking at clothes, head nodding, or some routine action – in a trance-like manner, and you cannot get their attention to stop it. Such “motor loops” can correspond to complex partial seizures.
- Abnormal facial movements or jerks: Brief, involuntary twitching of the face, eyes rolling upward, slight jerking of one side of the body, or eyelid fluttering can be seizures in young children. These movements might last only a second or two. In babies, even subtle eye or mouth movements (like rhythmic sucking or chewing motions unrelated to feeding) can be seizure equivalents.
- Sudden arrest of speech or activity: A child who is talking or playing may abruptly halt and freeze for a moment, possibly with a blank look, then resume. This could be an atypical absence or focal seizure manifesting as a brief arrest in activity. In benign rolandic epilepsy, seizures often occur at night, but if they happen while awake, a classic sign is the child unable to speak or producing only garbled sounds for a brief period (an ictal speech arrest) despite being awake and understanding.
- Regression or plateau in development: One “silent” clue to epileptic activity is an unexplained loss of developmental milestones or a halt in progress. For example, a toddler who was starting to say words may lose those words, or a potty-trained child starts having accidents again (seizures can cause urinary incontinence). A gradual deterioration in behavior, attention, or speech with no other obvious cause may warrant an EEG to check for subclinical seizures.
- Night-time disturbances and sleep issues: Many pediatric seizures occur in sleep. A child who frequently wakes up confused, screaming (night terrors aside), sweating, or is found in unusual positions could be having nocturnal seizures. Also, unusual sleep patterns – such as needing excessive sleep or being oddly tired in the morning despite a full night’s rest – might hint that the brain’s sleep is being disrupted by epileptic activity. Some children with ESES, for instance, have very restless sleep or parasomnias.
- Unexplained behavior changes or “daytime spells”: Episodes of sudden fear, panic, or laughing without reason, sudden rage or confusion, or “spacing out” in the middle of an activity can all be seizure manifestations in young kids. If a typically active child suddenly has moments of floppiness or head-dropping (losing muscle tone briefly), those could be atonic seizures. In general, erratic behavior that is “off” with no clear trigger may warrant neurological evaluation.
Being aware of these subtleties is important. Often it is the parents’ careful observations (e.g. keeping a seizure diary, recording events on video) that help doctors differentiate true seizures from normal behavior or other issues. If such signs are present, an EEG should be done, and even if the routine awake EEG is normal, a sleep EEG or overnight study may be necessary. In the context of a child with developmental delays, catching previously “silent” seizures can be a turning point for getting proper treatment.
Treatment Considerations: Should Spikes Be Treated if Seizures Are Absent?
Treating an epileptic disorder usually aims to stop clinical seizures, but what if a child’s only “symptom” is the abnormal EEG activity? This is a contentious area in pediatric neurology. On one hand, there is concern that frequent spikes (especially if they amount to an ESES pattern) can cause “transient cognitive impairments” and cumulatively derail development. Indeed, studies have shown that a higher load of interictal spikes correlates with lower IQ and worse memory/executive function in children. Children with very frequent discharges can have momentary lapses in attention or memory when a spike occurs, even if no overt seizure happens – this has been demonstrated by neuropsychological tests during EEG monitoring. Over months and years, an unchecked spike-wave encephalopathy could contribute to ADHD-like symptoms, learning disabilities, or language problems. Given this, many experts argue that treating the EEG activity itself (even in the absence of obvious seizures) might improve cognitive outcomes. This is the rationale for aggressively treating conditions like LKS and CSWS with medications or other therapies as soon as they are identified.
On the other hand, medications for epilepsy have side effects, and not all EEG spikes are equally harmful. Some children with a modest number of spikes may develop normally and never have a seizure, so exposing them to medication might be unnecessary. The decision often comes down to whether the child is experiencing developmental or behavioral problems that could be attributed to the EEG discharges. If a child is completely asymptomatic and thriving, many clinicians would opt to observe rather than medicate immediately. For example, in benign rolandic epilepsy, if seizures are infrequent, mild, and occur only at night, and the child is near the typical age of remission, one might choose not to start daily anti-epileptic drugs (AEDs). Clinical guidelines note that children with rolandic spikes “may not need antiepileptic medication, particularly if seizures are infrequent, nocturnal, or the child is close to remission age”. Conversely, if that child had frequent daytime seizures or notable learning difficulties, treatment would be recommended.
In syndromes like LKS/CSWS, treatment is almost always pursued because the risk of permanent language or cognitive loss is high without intervention. The goal in those cases is to suppress the epileptiform discharges (especially during sleep) as much as possible, to allow the brain to resume normal development. Valproate (valproic acid) is a common first-line medication in these situations. It’s a broad-spectrum AED that has been used for decades in generalized and focal epilepsies, and it can reduce both seizures and interictal spikes. In fact, the most commonly used drugs for CSWS/ESES in clinical practice are valproate, ethosuximide, and levetiracetam, alone or in combination. Benzodiazepines (like clobazam or high-dose diazepam at night) and corticosteroids (like high-dose prednisone or ACTH) are also frequently used if needed. There is some evidence supporting this approach: for example, Inutsuka et al. (2006) reported that using high-dose valproate (sometimes combined with ethosuximide) led to long-lasting seizure control and partial recovery of cognitive function in 10 of 15 children (67%) with CSWS. However, results vary – another series found that valproate alone was ineffective in many CSWS patients, and often a multi-modal approach or alternative therapies (like steroids or even epilepsy surgery if a focal lesion is driving the discharges) are required in refractory cases. Despite mixed study results, a trial of therapy is usually warranted when a child has clear cognitive or language regression associated with frequent EEG spikes. The treating neurologist will carefully monitor whether suppressing the discharges (which can be seen by repeat EEGs) correlates with improvement in the child’s function – essentially an N-of-1 trial in each patient.
Even outside of well-defined syndromes, there is growing interest in treating subclinical epileptiform EEG activity in children with developmental disorders. Some controlled trials have found that reducing interictal discharges with medication can lead to behavioral and learning improvements. For instance, both valproate and lamotrigine (another broad-spectrum AED) were shown in trials to decrease the burden of epileptiform EEG activity and improve behavior in children who had epilepsy with cognitive problems. This suggests that at least in some cases, the spikes themselves were contributing to the children’s developmental issues, and quieting them helped the brain function better. Still, it’s important to approach this on a case-by-case basis. Current expert reviews emphasize that there is only limited evidence proving long-term benefit from treating interictal spikes per se. Thus, many neurologists will only treat EEG abnormalities proactively if there is a strong suspicion that they are causing harm (e.g. clear regression, or an epilepsy syndrome known to cause such regression). Families should be counseled about the uncertain benefits and potential side effects. In summary, if a child has developmental stagnation or regression and frequent EEG spikes, a trial of anti-epileptic medication is often justified, whereas a child who is completely normal except for EEG might just be observed with periodic EEGs. This nuanced approach attempts to balance the possible upside of therapy against the risks.
Success Stories: Improving Development by Treating “Silent” Epilepsy
Although treating EEG spikes without obvious seizures is somewhat controversial, there are remarkable anecdotes and small studies that fuel optimism. One notable case series reported by neurologist J. Gordon Millichap described three young children (ages 3, 4, and 5) who had autism, were non-verbal, and had epileptiform EEG discharges, though none had a history of clinical seizures. These children were started on low-dose valproic acid (125 mg three times daily). Within one month of treatment, their language and social skills showed dramatic improvement, to the point that they no longer met the diagnostic criteria for autism. The improvement was sustained on follow-ups 7–11 months later. In other words, suppressing the subclinical seizure activity appeared to “release” the children’s developmental potential, allowing them to rapidly acquire language and social engagement that had previously been stalled. The author of that report emphasized the importance of doing sleep EEGs in children with autism and regression, as standard awake EEGs could miss these epileptiform bursts. These cases, while few, provide a proof-of-concept that sometimes what looks like primary autism or developmental delay can actually be (or be exacerbated by) an occult epileptic process – one that is at least partially reversible with therapy.
Another true story can be inferred from the scenario that inspired this article: a 3-year-old child who was non-verbal and initially suspected to have autism (or another developmental disorder) was found to have frequent EEG spikes during sleep. The child did not meet full criteria for LKS (perhaps because there was no clear loss of language – the child hadn’t spoken yet at all) and did not have the classic extreme spike burden of ESES, and MRI imaging was normal (ruling out focal cortical dysplasia or other lesions). In essence, it was an idiopathic epileptic encephalopathy without a name – a subclinical epilepsy impairing the child’s ability to develop speech. The treating clinicians decided to try antiseizure medication despite the lack of obvious seizures. Valproate was chosen (as it was in the above autism cases) due to its broad efficacy and safety profile in young children. Over the ensuing months, the parents witnessed a transformation: the child began to gain words and make cognitive strides that had not occurred before. This kind of outcome, though not guaranteed, is incredibly validating. It suggests that the pathological EEG spikes were indeed a barrier to normal brain function – akin to “electrical noise” drowning out the brain’s signals for learning and communication – and that reducing this noise pharmacologically allowed the child’s innate developmental programs to resume.
It’s worth noting that such children may fall into what some researchers call the “epilepsy aphasia spectrum,” which includes milder variants of LKS/CSWS. There is a continuum from benign rolandic epilepsy (which can have very focal spikes and minimal impact) through intermediate forms to severe LKS/CSWS. Children can move along this spectrum. In our hypothetical case, while the child wasn’t “classical” LKS, treating the spikes early may have actually prevented progression into a more severe syndrome. Differentiating these conditions formally may be less important than recognizing the shared feature: epileptiform EEG activity that interferes with neurodevelopment. The key takeaway for doctors and parents is to keep an open mind – if a child has unexplained developmental delay or regression, obtaining an overnight EEG can be very informative, and if significant abnormalities are found, a therapeutic trial might be life-changing.
Of course, not all cases have such positive outcomes. Some children show no improvement despite spike-suppressing treatments, and some partially improve but not to normal levels. There is also the natural course to consider: for instance, in CSWS, even if we do nothing, the EEG will eventually normalize by the teenage years in many cases – but we risk the child losing precious years of learning in the interim. Therefore, most practitioners err on the side of treatment in impactful syndromes. The success stories like the above provide hope and underscore why researchers are studying this area intensely. They also highlight why multidisciplinary collaboration (neurologists, epileptologists, neuropsychologists, speech therapists) is important: improvements in EEG and medication should be paired with therapies (speech/language therapy, behavioral interventions) to maximize the child’s gains once the “brain fog” lifts.
Why Spikes Occur Despite a Negative MRI: The Puzzle of Idiopathic Epileptiform Activity
It can be perplexing for parents to hear that their child’s MRI brain scan is normal, and yet the EEG is markedly abnormal. MRI is excellent at revealing structural problems – tumors, malformations, scars, etc. – but epileptiform discharges can arise from more subtle dysfunctions that MRI cannot see. Think of the brain as an electrical circuit: MRI can show if any wires are broken or misplaced, but it won’t show if the circuitry is misfiring. In many childhood epilepsy syndromes, the issue is one of network excitability rather than a gross lesion. For example, benign centrotemporal spikes are thought to reflect a transient imbalance in the developing brain, possibly due to genetic factors that slightly alter neuronal excitability or connectivity. The child’s brain is structurally intact; it’s just running a somewhat “noisy” operating system for a few years until it outgrows the issue. As noted, rolandic epilepsy has a genetic basis with evidence of heritability (family members might have had febrile seizures or similar EEG patterns), but no single gene is determinative – it appears to be multifactorial and related to brain maturation. The concept of a “maturational derangement” suggests that the timing of brain network development is slightly off in these kids, producing spikes that eventually disappear when maturation catches up.
In epileptic encephalopathies like CSWS, research indicates a dysfunction in the thalamus-cortex network that underlies sleep rhythms. The thalamus acts as a pacemaker for brain rhythms during sleep; if an early-life insult (like a microscopic injury or even a neurodevelopmental abnormality too subtle for MRI) perturbs that system, the result can be pathological oscillations (spike-waves) instead of normal sleep spindles. In roughly half of CSWS cases a lesion is identified (often a prior stroke or a cortical dysplasia in a strategic location), but in the rest, the cause might be a “functional lesion” – meaning an abnormal circuit that doesn’t show up anatomically. There could also be unknown genetic contributors; interestingly, grin2a mutations are not only in LKS but also found in some CSWS cases, indicating a possible common pathway involving glutamate neurotransmission. Additionally, some idiopathic cases might actually have ultra-microscopic malformations or dysplasias that our current MRI technology just can’t resolve. The take-home message is that a normal MRI does not equate to a normal brain electrophysiology. It’s quite possible to have significant electrical disturbances with a clean scan.
From a clinical standpoint, when MRI is normal, doctors look harder for other clues: metabolic or genetic tests might be done to see if there’s an inborn error or mutation causing the epilepsy. Often none are found, and the epilepsy is labeled “cryptogenic” or “idiopathic.” This can be frustrating, but also somewhat reassuring – at least there is no tumor or degenerative disease. The reason for the spikes might be hidden in the genome or simply in how that child’s brain networks wired themselves during development. As science advances, we may identify more subtle biomarkers (e.g. PET scans or MEG scans sometimes show abnormalities where MRI is normal). For now, treatment in MRI-negative cases is guided by the EEG phenotype and clinical picture. Whether or not we know the precise cause, if a child has an EEG pattern like CSWS or LKS, we treat it similarly to cases with known causes.
Interestingly, even truly benign centrotemporal spikes tell us something about the child’s brain: studies have noted that children with rolandic spikes – even those who never have seizures – can have higher rates of mild developmental concerns (like speech sound disorders or attention issues) than children with completely normal EEGs. The spikes may be a marker of a brain that is “wired differently” in some minor way. That said, long-term follow-ups show these children do well – the spikes vanish by the teenage years and any early learning issues usually resolve. This reinforces that such spikes are benign variants of brain activity, not sustained pathologies. The reason they cluster in childhood (4–14 years) is likely because the brain, in its plastic developing state, is prone to oscillatory instability that naturally stabilizes with maturation.
In summary, EEG spikes without MRI findings often point to a network instability of the developing brain. Whether it’s due to genetic, microscopic cortical organization quirks, or immune factors, metabolic or any other factors, we often cannot say. But we do know many of these conditions are age-related: the brain “outgrows” the tendency (as in benign focal epilepsies and even in CSWS which is self-limited to childhood). The key is managing the condition during that vulnerable window to mitigate any harm.
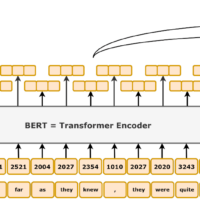
How AI and LLM Technology Can Aid Diagnosis and Therapy
The challenges posed by these pediatric EEG disorders – subtle signs, complex EEG interpretations, and decision-making about when to treat – make them ripe for assistance from advanced computational tools. Artificial intelligence (AI), particularly machine learning and deep learning, is increasingly being applied in neurology to improve diagnosis and personalize treatment. For EEG analysis, AI algorithms can be trained to recognize epileptiform patterns with high sensitivity. For example, researchers have developed deep learning models that automatically detect spikes and even quantify the spike-wave index (the percentage of sleep occupied by spikes) in children with CSWS. In one study, an AI system (dubbed SCORE-AI) was trained on over 30,000 clinical EEGs to classify them – it could distinguish normal vs. abnormal EEGs and further identify which abnormal ones had focal epileptiform discharges. Such tools could greatly help neurophysiologists by screening long EEG recordings (including overnight studies) and flagging periods of concern. This is especially useful for catching conditions like ESES early, where the timing and percentage of spikes matter; an AI can measure this far faster than a human reviewing hours of data.
AI is also being explored to predict outcomes – for instance, using machine learning on EEG and MRI data to predict which children with focal epilepsy might progress to ESES or which might benefit from surgery. For children who cannot express what they are feeling, AI might detect subtle autonomic changes or patterns in behavior that correlate with seizures (via wearable devices or video analysis). All of this can lead to earlier intervention.
Meanwhile, Large Language Models (LLMs) like GPT-4 (the AI behind ChatGPT) are showing potential in the medical domain as well. These models can rapidly digest and summarize vast amounts of medical literature and guidelines, which can help clinicians stay updated on rare conditions and emerging treatments. In epilepsy care, experts suggest that LLMs could be integrated to accelerate diagnosis and aid clinical decision-making. For example, an LLM could be used to input a detailed case description (including EEG findings, development history, etc.) and it might suggest a list of possible syndromes (like LKS, atypical autism with epileptiform EEG, etc.) for the neurologist to consider. One recent study even tested GPT-4 on the task of distinguishing epileptic seizures from psychogenic non-epileptic events based on patient histories – a nuanced diagnostic problem – and explored its performance. While LLMs are not yet as reliable as expert physicians for making diagnoses, the field is moving fast. They show promise in supporting clinicians by providing second opinions or summarizing patient data.
For families, LLM-based chatbots could provide accessible information and support. Imagine a parent whose child was just diagnosed with CSWS – they could ask a vetted medical chatbot questions 24/7 to better understand the condition, treatment options, or how to manage learning difficulties at home. Properly designed, these AI systems can break down barriers to information, helping parents and patients navigate the complex journey of epilepsy care. They might also assist in tracking a child’s progress: parents could report daily observations to an AI system that analyzes trends or flags concerning changes to notify the care team.
On the therapy side, AI and LLMs might contribute to personalized education plans and cognitive therapies. For example, AI-driven apps can adjust in real-time to a child’s cognitive level, providing brain training exercises that adapt if the child is having a “spiky” EEG day versus a good day. There is also interest in neurofeedback systems, where an AI could use EEG input to help train the child’s brain to suppress spikes (an experimental idea for the future).
It is important to temper enthusiasm with caution: neither AI nor LLMs replace the nuanced judgment of doctors or the need for direct patient evaluation. They are tools – and their outputs need validation. Ethical considerations (privacy of EEG data, ensuring medical accuracy of chatbot responses) are paramount. But if implemented wisely, these technologies hold great potential. They could ensure that no subtle seizure goes unnoticed in a long recording, and that clinicians have at their fingertips the collective knowledge of thousands of cases to inform treatment decisions. As one review noted, epilepsy care could benefit from integrating LLMs to streamline diagnosis, assist with patient counseling, and even suggest management plans (for example, recommending when to consider a steroid trial for ESES or a gene test for a child with LKS features).
In the near future, one can envision a scenario where a concerned mother describes her child’s nightly episodes to a digital health assistant; the system, drawing on a vast database, recognizes the red flags for possible CSWS and urges prompt EEG evaluation – potentially leading to an earlier diagnosis than might have occurred otherwise. Or a system where, after starting a medication like valproate, an AI analyzes follow-up EEGs and developmental assessments, then advises the physician whether enough improvement has been achieved or if further intervention is warranted.
In conclusion, the intersection of pediatric epilepsy, developmental disorders, and EEG is complex, but advances in both medical science and technology are providing hope. We are learning that some “benign” EEG patterns are truly benign, while others hide serious neurodevelopmental risks – making it crucial to discern which is which. We also see that even when seizures are not evident, treating the brain’s electrical storms can, in select cases, unlock a child’s ability to learn and grow. As research marches on, and with the aid of AI to augment our capabilities, we move closer to ensuring that every child with these conditions gets timely, effective care – maximizing their chances for a healthy, communicative life.
Sources: The information in this article is drawn from a range of current medical literature and case studies, including clinical guidelines on benign childhood epilepsies, research on the cognitive effects of epileptiform EEG discharges, and documented improvements in children treated with antiepileptic medication despite no overt seizures. The role of valproate and other therapies in conditions like ESES is based on clinical series and reviews. Signs of subtle seizures are noted per pediatric neurology resources. Discussions on LKS and CSWS reference definitions and findings from rare disease databases and epileptology texts. Finally, insights into AI and LLM applications in epilepsy care are informed by recent analyses in the field of neuroinformatics. Each citation corresponds to the relevant source material supporting the statements made.